
If you plan enough time for sleep each night but still feel tired during the day, there could be a reason besides sleep apnea or insomnia.

How do you know if you have narcolepsy? Persistent excessive daytime sleepiness (EDS) is an essential symptom.
You may have a narcoleptic episode in which you suddenly fall asleep with no warning. This can happen at any time during the day and last from several seconds to a few minutes.
Some people with narcolepsy experience brief and sudden loss of motor control, but not everyone with the condition has this symptom.
If you or someone you know has regular daytime sleepiness, identifying whether narcolepsy is the cause is an important step toward support and treatment.
Narcolepsy is a chronic neurological condition that affects the way the brain regulates sleep-wake cycles. It affects as many as
People can develop symptoms at any time in their lives, although more diagnoses happen around 15 and 36 years of age.
Your symptoms may depend on the type of narcolepsy you have.
Narcolepsy has five main symptoms, represented by the acronym CHESS. Not everyone with narcolepsy has all five symptoms, although excessive daytime sleepiness is the most common. In fact, according to the
For some, symptoms may fade or even disappear over time, as seen in a 10-year
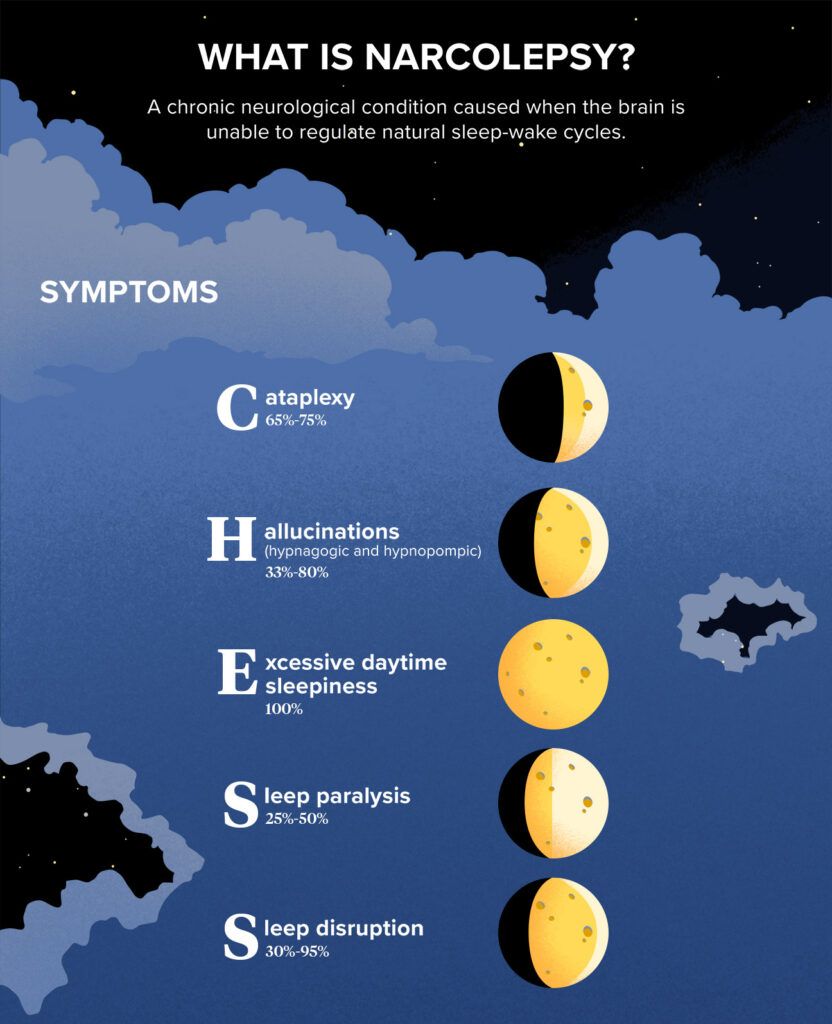
Cataplexy
This symptom is a short-lived and sudden loss of muscle tone that often happens with strong emotions like anger or laughter. It’s associated with type 1 narcolepsy only.
Cataplexy lasts only a few minutes and resolves on its own with no loss of consciousness.
The number of cataplexy attacks a person has can vary. While some people only have a few cataplexy attacks in their lives, others may experience several attacks in one day.
Cataplexy attacks can be mild and involve only minor muscle tone weakness. You may notice that your eyelids start drooping, or you have a slight facial twitch or flicker. Other mild symptoms include difficulty speaking, jaw tremors, or knee trembles.
If you have a severe cataplexy attack, your entire body can collapse, and you may not be able to speak, move, or keep your eyes open.
Hallucinations
These hallucinations are mainly visual and are frequently described as dream-like experiences during waking (hypnopompic) or falling asleep (hypnagogic). Images can be very vivid and sometimes scary. About
Excessive daytime sleepiness
All people with narcolepsy have this symptom, which is the most debilitating. People who have this symptom have difficulties staying alert or awake during the day.
This constant overwhelming sleepiness comes with “sleep attacks,” which can happen suddenly and often occur no matter how much sleep you’ve gotten.
You may find yourself falling asleep easily no matter where you are — at your desk at school or in your office, or even while driving. Some even report falling asleep mid-conversation.
You can fall asleep for a few seconds or a few minutes, aka microsleeps. Many find that short naps during the day can help them feel refreshed, if only for a short period.
Sleep paralysis
This is characterized by a feeling of being unable to move during the transition between wakefulness and sleep.
This usually lasts only a few seconds to minutes, and people usually can move and speak once the episode ends. About
Sleep disruption
Many people wake up during the night. This is not uncommon.
But when you have narcolepsy, you may find yourself repeatedly waking up throughout the night or even wide awake for most of it.
This is another common symptom of narcolepsy, with an estimated
Other common symptoms of narcolepsy
In addition to the primary five symptoms of narcolepsy, you can also experience these other symptoms:
- fragmented sleep and insomnia: sleep interrupted by vivid dreams, sleep apnea, insomnia, restless leg movements, and acting out while in a dream state
- automatic behaviors: automatically continuing an activity (such as eating, talking, typing, or driving) for a few seconds or minutes after dropping off to sleep without even being aware you’re doing it
Narcolepsy symptoms in children may not look the same. Some symptoms are shared between adults and children, but children can experience other effects like irritability and restlessness.
Childhood narcolepsy symptoms can often be mistaken for behavioral issues with other causes. Some medical professionals believe that true childhood narcolepsy rates are higher than reported.
Some research suggests that children with narcolepsy experience cataplexy more than adults, although it’s milder and often resembles a facial tic. Cataplexy in children may not have the trigger of strong emotions, although this can change with time.
Researchers know more about the cause of type 1 narcolepsy than they do about type 2.
Type 1 involves a loss of the brain cells that produce hypocretin, aka orexin, which influences how you sleep.
Some people with type 1 experience a hypocretin cell reduction as high as 80% to 90%. Without enough hypocretin, the brain can’t properly regulate sleep and wake cycles. Hypocretin is an essential neurotransmitter in the brain for being alert and wakeful during the day.
Research has not yet identified the exact cause of type 2 narcolepsy. This type of narcolepsy isn’t associated with reduced hypocretin or cataplexy like type 1. Some instances of type 2 have been reported after viral infections, but more research is needed to understand the cause of this type of narcolepsy.
Contributing factors can increase the chance of developing narcolepsy. These include:
- genetics such as the human leukocyte antigen,
HLA DQB1*06:02 - autoimmune disorders
- family history – first degree relatives of a person with type 1 narcolepsy are
10 to 40 times more likely than the general population to have narcolepsy
Some factors can contribute to an autoimmune response that may trigger narcolepsy or cause symptoms. Examples of these include:
- stress
- a changing sleep schedule
- a bacterial or viral infection
- flu season
- hormonal fluctuations
If you think you may have narcolepsy, consider reaching out to a medical doctor. They will assess your symptoms and medical history and determine whether narcolepsy is the cause or some other medical condition.
They will likely recommend a sleep test at night to rule out any other sleep disorders. Then, they may recommend a napping studying in the morning to access daytime sleepiness. Additional laboratory testing may be done after a diagnosis of narcolepsy to determine whether you have type 1 or type 2 narcolepsy.
Treatment for narcolepsy may include medications — such as stimulants and alerting antidepressants — to help ease symptoms, behavior therapy, or counseling.
Lifestyle changes — such as scheduling short naps during the day — may also be helpful.
You can also find help and support by joining a narcolepsy support group. You can find an online support group or one near you by visiting the following: